Respirology | APSR Respiratory Medicine Journal

Invalid Access Key! Invalid Access Key!
| Posted: 28 Feb 2021 05:00 AM PST  Clinical trials with NULIBRY or recombinant cPMP showed a meaningful increase in overall survival compared to a natural history studyNULIBRY is BridgeBio's first FDA-approved therapeuticPALO ALTO, Calif., Feb. 28, 2021 (GLOBE NEWSWIRE) -- BridgeBio Pharma, Inc. (Nasdaq: BBIO) (BridgeBio) and affiliate Origin Biosciences, Inc. (Origin) today announced the U.S. Food and Drug Administration (FDA) has approved NULIBRY™ (fosdenopterin) for Injection as the first therapy to reduce the risk of mortality in patients with molybdenum cofactor deficiency (MoCD) Type A. This is the first therapy of its kind. The novel therapy was developed based on BridgeBio's commitment to developing a treatment for MoCD Type A in collaboration with the experts and families in the MoCD Type A community. The announcement was made on Rare Disease Day, which aims to raise awareness about the impact of rare diseases on patients.MoCD Type A is an ultra-rare and progressive condition, known to impact less than 150 patients globally with a median survival of four years. MoCD Type A presents shortly after birth, often with severe encephalopathy and intractable seizures. NULIBRY is a first-in-class approved cPMP substrate replacement therapy."The FDA's approval of NULIBRY means that patients with MoCD Type A and their families have an approved therapy for the first time. It also reflects our belief that every life matters and that no disease is too rare to address. As is often true in rare disease drug development, this was a community effort in which we were able to play a part – we'd like to thank the patients, caregivers, physicians, scientists, and advocates who played an essential role in achieving this important milestone," said BridgeBio founder and CEO Neil Kumar, Ph.D.The efficacy of NULIBRY for the treatment of patients with MoCD Type A was established based on data from three clinical trials compared to data from a natural history study. In these studies, NULIBRY or recombinant cPMP (rcPMP; same active moiety and biologic activity as NULIBRY) reduced the risk of death by 82% compared to the untreated, genotype-matched, historical control group in the natural history study (HR=0.18, 95% CI 0.04, 0.72). At three years on study, the probability of survival in NULIBRY or rcPMP-treated patients (n=13) was 84% (95% CI 49%,96%) compared to 55% (95% CI 30%,74%) for untreated genotype-matched patients in the historical control group (n=18) at three years (Figure 1). In addition to the survival analysis, treatment with NULIBRY led to a reduction of urine concentrations of S-sulfocysteine (SSC), a toxic substance that leads to neurological damage, in patients with MoCD Type A, and the reduction was sustained with long-term treatment over 48 months.Animal studies have identified that NULIBRY has phototoxic potential. In the clinical trials, the most common adverse reactions reported in two or more Nulibry-treated patients with MoCD Type A were catheter-related complications (89%), pyrexia (fever; 78%), viral infection (56%), pneumonia (44%), otitis media (ear infection; 44%), vomiting (44%) and cough/sneezing (44%). Adverse reactions for the rcPMP-treated patients were similar to the NULIBRY-treated patients."Today's approval represents new hope for patients and families affected with MoCD Type A," said Donald Basel, M.D., section chief and associate professor of pediatrics at Children's Wisconsin. "I am encouraged by the clinical data showing that NULIBRY not only lowers the levels of toxic SSC, but importantly extends the lives of patients who previously had only a three- to four-year median survival."NULIBRY was reviewed under Priority Review and received Orphan Drug Designation, Breakthrough Therapy Designation and Rare Pediatric Disease Designation from the FDA. With this approval, the FDA also issued a Rare Pediatric Disease Priority Review Voucher (PRV) to Origin.BridgeBio and Origin are committed to patient access and have developed a comprehensive patient support program, ForgingBridges, to help patients access NULIBRY. ForgingBridges also provides tools and resources to help patients and caregivers throughout their treatment journey with NULIBRY.Visit NULIBRY.com for more information, including full Prescribing Information.About Molybdenum Cofactor Deficiency (MoCD) Type A MoCD Type A is an autosomal recessive, inborn error of metabolism caused by mutations in the molybdenum cofactor synthesis 1 gene (MOCS1) and characterized by a deficiency in molybdenum cofactor (MoCo) production, leading to a lack of molybdenum-dependent enzyme activity1,2. The lack of activity leads to decreased sulfite oxidase activity with buildup of sulfite and secondary metabolites (such as S-sulfocysteine (SSC)) in the brain, which causes irreversible neurological damage.2MoCD Type A is an ultra-rare disease. The incidence and prevalence of MoCD Type A in the United States are not known, but the estimated incidence is 1 per 342,000 to 411,000 live births (0.24 and 0.29 per 100,000).3 Based on these estimates, MoCD Type A is likely to be underdiagnosed, with an estimated 22 to 26 missed diagnoses per year in the United States and European Union.The most common presenting symptoms of MoCD Type A are seizures, feeding difficulties and encephalopathy. Patients with MoCD Type A who survive beyond infancy typically suffer from progressive brain damage, which presents in characteristic patterns on magnetic resonance imaging (MRI). This damage leads to severe psychomotor impairment and an inability to make coordinated movements or communicate with their environment.About NULIBRY (fosdenopterin) for Injection NULIBRY (fosdenopterin) for Injection is a substrate replacement therapy that provides an exogenous source of cPMP, which is converted to molybdopterin. Molybdopterin is then converted to molybdenum cofactor, which is needed for the activation of molybdenum-dependent enzymes, including sulfite oxidase, an enzyme that reduces levels of neurotoxic sulfites. It is the first and only FDA-approved therapy indicated to reduce the risk of mortality in patients with MoCD Type A, and clinical trials have demonstrated that patients treated with NULIBRY or rcPMP had an improvement in overall survival compared to the untreated, genotype-matched, historical control group.IMPORTANT SAFETY INFORMATIONWARNINGS AND PRECAUTIONSPotential for Photosensitivity NULIBRY can make the patient oversensitive to sunlight. NULIBRY-treated patients or their caregivers are advised to avoid or minimize patient exposure to sunlight and artificial UV light and adopt precautionary measures when exposed to the sun, including wearing protective clothing and sunglasses, and use broad-spectrum sunscreen with high SPF in patients 6 months of age and older. If photosensitivity occurs, caregivers/patients are advised to seek medical attention immediately and consider a dermatological evaluation.ADVERSE REACTIONS The most common adverse reactions in NULIBRY-treated patients were infusion catheter–related complications (89%), pyrexia (fever) (78%), viral infection (56%), pneumonia (44%), otitis media (ear infection) (44%), vomiting (44%), and cough/sneezing (44%). Adverse reactions for rcPMP-treated patients were similar to the NULIBRY-treated patients.PATIENT COUNSELING INFORMATION Please read the FDA-approved NULIBRY Prescribing Information and Instructions for Use and follow the instructions on how to prepare and administer NULIBRY.NULIBRY has a potential for photosensitivity; see Warnings and Precautions. Seek medical attention immediately if the patient develops a rash or if they notice symptoms of photosensitivity reactions (redness, burning sensation of the skin, blisters).You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088.Clinical Trials4 The efficacy of NULIBRY for the treatment of patients with MoCD Type A was established based on data from three clinical trials compared to data from a natural history study.Study 1: Study 1 was a prospective, open-label, single-arm, dose-escalation study in patients with MoCD Type A who were receiving treatment with rcPMP prior to treatment with NULIBRY. Study 1 included eight patients, six of whom previously participated in Study 3. The initial NULIBRY dosage was matched to the patient's rcPMP dosage upon entering the study. The NULIBRY dosage was then titrated over a period of five months to a maximum dosage of 0.9 mg/kg administered once daily as an intravenous infusion.Study 2: Study 2 was a prospective, open-label, single-arm, dose-escalation study in one patient with confirmed MoCD Type A who had not been previously treated with rcPMP. The initial dosage of NULIBRY in Study 2 was based on the gestational age of the patient (i.e., 36 weeks). The initial dosage was then incrementally escalated up to a maximum dosage of 0.98 mg/kg administered once daily as an intravenous infusion (1.1 times the maximum approved recommended dosage).Study 3: Study 3 was a retrospective, observational study that included 10 patients with a confirmed diagnosis of MoCD Type A who received rcPMP. Six of these 10 patients were later enrolled in Study 1 to receive treatment with NULIBRY.NULIBRY Efficacy and Safety Data4 The efficacy of NULIBRY and rcPMP were assessed in a combined analysis of the 13 patients with genetically confirmed MoCD Type A from Study 1 (n=8), Study 2 (n=1), and Study 3 (n=4), who received substrate replacement therapy with NULIBRY or rcPMP.Of the 13 treated patients included in the combined analysis, 54% were male, 77% were White and 23% were Asian; the median gestational age was 39 weeks (range 35 to 41 weeks). Of these 13 treated patients, the age at first dose was ≤ 14 days for 10 patients (with five patients initiating treatment at one day of age) and ≥32 days and <69 days for the remaining three patients.Efficacy was assessed by comparing overall survival in pediatric patients treated with NULIBRY or rcPMP (n=13) with an untreated natural history cohort of pediatric patients with genetically confirmed MoCD Type A who were genotype-matched to the treated patients (n=18). Patients treated with NULIBRY or rcPMP had an improvement in overall survival compared to the untreated, genotype-matched, historical control group. Results were similar when comparing treated patients with all patients in the untreated natural history cohort with genetically confirmed MoCD Type A (n=37, including the 18 genotype-matched untreated patients as well as 19 additional untreated patients who were not genotype-matched).Treatment with NULIBRY resulted in a reduction of urine concentrations of SSC in patients with MoCD Type A, and the reduction was sustained with long-term treatment over 48 months. The baseline level of urinary SSC normalized to creatinine was characterized in one patient (Study 2) with a value of 89.8 umol/mmol. Following treatment with NULIBRY in Studies 1 and 2 (n=9), the mean±SD levels of urinary SSC normalized to creatinine ranged from 11 (±8.5) to 7 (±2.4) umol/mmol from month 3 to month 48.The safety of NULIBRY was assessed in 37 pediatric patients and healthy adults who received at least one intravenous infusion of NULIBRY or rcPMP. Of these 37 patients/healthy adults, 13 were pediatric patients with MoCD Type A in Studies 1, 2 and 3; six were pediatric patients with presumptive MoCD Type A but who were later confirmed to not have MoCD type A; and 18 were healthy adults (without MoCD Type A) in a Phase 1 study. The most common adverse reactions from NULIBRY-treated patients in Studies 1 and 2 were catheter-related complications, pyrexia (fever), viral infection, pneumonia, otitis media (ear infection), vomiting and cough/sneezing. Adverse reactions for the rcPMP-treated patients from Study 3 were similar to the NULIBRY-treated patients, with the exception of the following additional adverse reactions that were reported in more than one patient: sepsis, oral candidiasis, varicella, fungal skin infection and eczema.About Origin Biosciences, Inc. Origin Biosciences, Inc., an affiliate of BridgeBio Pharma, Inc., is a biotechnology company that developed and commercialized NULIBRY for the treatment of patients with a diagnosis or presumptive diagnosis of MoCD Type A. Origin Biosciences, Inc. is led by a team of veteran biotechnology executives. For more information on Origin Biosciences, Inc., please visit the company's website at www.origintx.com.About BridgeBio Pharma, Inc. BridgeBio is a team of experienced drug discoverers, developers and innovators working to create life-altering medicines that target well-characterized genetic diseases at their source. BridgeBio was founded in 2015 to identify and advance transformative medicines to treat patients who suffer from Mendelian diseases, which are diseases that arise from defects in a single gene, and cancers with clear genetic drivers. BridgeBio's pipeline of over 30 development programs includes product candidates ranging from early discovery to late-stage development. For more information visit bridgebio.com.BridgeBio Pharma Forward-Looking Statements This press release contains forward-looking statements. Statements we make in this press release may include statements that are not historical facts and are considered forward-looking within the meaning of Section 27A of the Securities Act of 1933, as amended (the Securities Act), and Section 21E of the Securities Exchange Act of 1934, as amended (the Exchange Act), which are usually identified by the use of words such as "anticipates," "believes," "estimates," "expects," "intends," "may," "plans," "projects," "seeks," "should," "will," and variations of such words or similar expressions. We intend these forward-looking statements to be covered by the safe harbor provisions for forward-looking statements contained in Section 27A of the Securities Act and Section 21E of the Exchange Act, and are making this statement for purposes of complying with those safe harbor provisions. These forward-looking statements, including statements relating to: the development by Origin Biosciences, Inc. (Origin) of NULIBRY™ (fosdenopterin) for Injection as the first therapy approved by the U.S. Food and Drug Administration (FDA) to reduce the risk of mortality in patients with molybdenum cofactor deficiency (MoCD) Type A; the potential for NULIBRY as the first and only FDA approved therapy for MoCD Type A; the efficacy of NULIBRY for the treatment of patients with MoCD Type A; the safety profile of NULIBRY for the treatment of patients with MoCD Type A, including the most common adverse reactions; BridgeBio's belief that every life matters and that no disease is too rare to address; the potential for NULIBRY to lower the levels of toxic S-sulfocysteine (SSC) and extend the lives of treated patients; the ability of NULIBRY to retain Orphan Drug Designation, Breakthrough Therapy Designation and Rare Pediatric Disease Designation from the FDA; the ability of ForgingBridges to be a comprehensive patient support program and help patients access NULIBRY; plans for the supply, manufacturing and distribution of NULIBRY; the incidence and prevalence of MoCD Type A; the current FDA-approved NULIBRY Prescribing Information and Instructions for Use; the planned approval of NULIBRY by foreign regulatory authorities and the necessary clinical trial results, and timing and completion of regulatory submissions related thereto; and the competitive environment and clinical and therapeutic potential of NULIBRY, reflect our current views about our plans, intentions, expectations, strategies and prospects, which are based on the information currently available to us and on assumptions we have made. Although we believe that our plans, intentions, expectations, strategies and prospects as reflected in or suggested by those forward-looking statements are reasonable, we can give no assurance that the plans, intentions, expectations or strategies will be attained or achieved. Furthermore, actual results may differ materially from those described in the forward-looking statements and will be affected by a variety of risks and factors that are beyond our control including, without limitation: the fact that there has never been an approved therapy for MoCD Type A; the safety, tolerability and efficacy profile of NULIBRY observed to date may change adversely in ongoing analyses of trial data or subsequent to commercialization; despite having ongoing interactions with the FDA or other regulatory agencies, the FDA or such other regulatory agencies may not agree with Origin's regulatory approval strategies, components of our filings, such as clinical trial designs, conduct and methodologies, or the sufficiency of data submitted; Origin may encounter delays in meeting manufacturing or supply timelines or disruptions in its distribution plans for NULIBRY; whether and when any regulatory submissions may be filed in various foreign jurisdictions and ultimately approved by foreign regulatory authorities; and potential adverse impacts due to the global COVID-19 pandemic such as delays in regulatory review, manufacturing and clinical trials, supply chain interruptions, adverse effects on healthcare systems and disruption of the global economy; as well as those set forth in the Risk Factors section of BridgeBio Pharma, Inc.'s most recent Annual Report on Form 10-K filed with the U.S. Securities and Exchange Commission (SEC) and in subsequent SEC filings, which are available on the SEC's website at www.sec.gov. Except as required by law, each of BridgeBio and Origin disclaims any intention or responsibility for updating or revising any forward-looking statements contained in this press release in the event of new information, future developments or otherwise. Moreover, BridgeBio and Origin operate in a very competitive environment in which new risks emerge from time to time. These forward-looking statements are based on each of BridgeBio's and Origin's current expectations, and speak only as of the date hereof.References 1Mechler K et al. Genet Med. 2015;17(12):965-970. 2Schwarz G. Cur Op in Che Bio. 2016;31:179-187. 3Mayr SJ, et al. Forecasting the incidence of rare diseases: an iterative computational and biochemical approach in molybdenum cofactor deficiency type A. Presented at the 2019 SSIEM meeting; September 3-6, 2019; Rotterdam, The Netherlands. 4NULIBRY (fosdenopterin) Origin Biosciences, Palo Alto, CA, USA; February 2021.NOTE: NULIBRY is a trademark of Origin Biosciences, Inc. Origin Biosciences, Inc. is a member of the BridgeBio family. ForgingBridges is a trademark of BridgeBio. Grace Rauh grace.rauh@bridgebio.com (917) 232-5478Source: BridgeBio Pharma, Inc.A Media Snippet accompanying this announcement is available by clicking on the image or link below: |
| Posted: 24 Feb 2021 06:50 AM PST 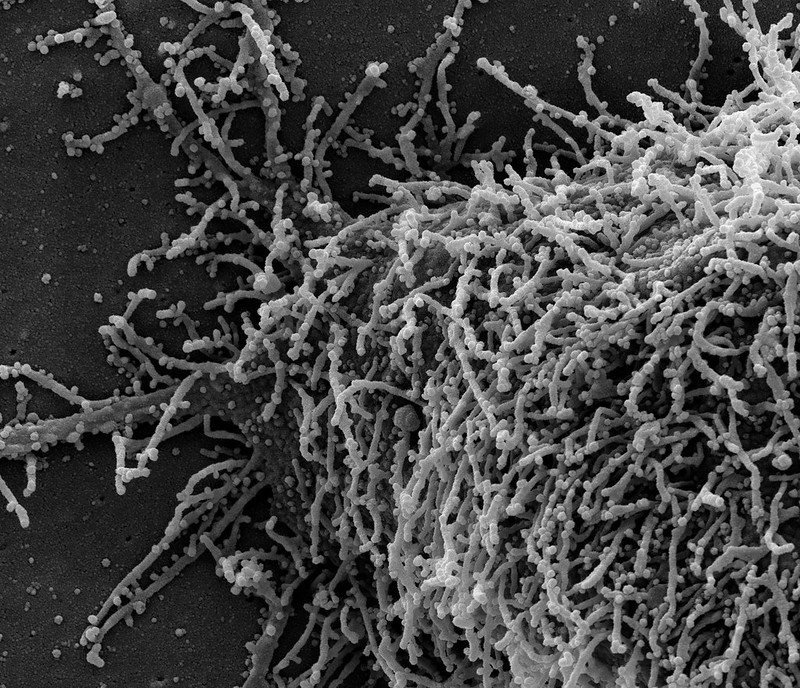 News Release Wednesday, February 24, 2021 People who have had evidence of a prior infection with SARS-CoV-2, the virus that causes COVID-19, appear to be well protected against being reinfected with the virus, at least for a few months, according to a newly published study from the National Cancer Institute (NCI). This finding may explain why reinfection appears to be relatively rare, and it could have important public health implications, including decisions about returning to physical workplaces, school attendance, the prioritization of vaccine distribution, and other activities. For the study, researchers at NCI, part of the National Institutes of Health, collaborated with two health care data analytics companies (HealthVerity and Aetion, Inc.) and five commercial laboratories. The findings were published on Feb. 24 in JAMA Internal Medicine. "While cancer research and cancer care remain the primary focus of NCI's work, we were eager to lend our expertise in serological sciences to help address the global COVID-19 pandemic, at the request of Congress," said NCI Director Norman E. "Ned" Sharpless, M.D., who was one of the coauthors on the study. "We hope that these results, in combination with those of other studies, will inform future public health efforts and help in setting policy." "The data from this study suggest that people who have a positive result from a commercial antibody test appear to have substantial immunity to SARS-CoV-2, which means they may be at lower risk for future infection," said Lynne Penberthy, M.D., M.P.H., associate director of NCI's Surveillance Research Program, who led the study. "Additional research is needed to understand how long this protection lasts, who may have limited protection, and how patient characteristics, such as comorbid conditions, may impact protection. We are nevertheless encouraged by this early finding." Antibody tests — also known as serology tests — detect serum antibodies, which are immune system proteins made in response to a specific foreign substance or infectious agent, such as SARS-CoV-2. This study was launched in an effort to better understand whether, and to what degree, detectable antibodies against SARS-CoV-2 protect people from reinfection with the virus. Working with HealthVerity and Aetion, NCI aggregated and analyzed patient information collected from multiple sources, including five commercial labs (including Quest Diagnostics and Labcorp), electronic medical records, and private insurers. This was done in a way that protects the privacy of an individual's health information and is compliant with relevant patient privacy laws. The researchers ultimately obtained antibody test results for more than 3 million people who had a SARS-CoV-2 antibody test between Jan. 1 and Aug. 23, 2020. This represented more than 50% of the commercial SARS-CoV-2 antibody tests conducted in the United States during that time. Nearly 12% of these tests were antibody positive; most of the remaining tests were negative, and less than 1% were inconclusive. About 11% of the seropositive individuals and 9.5% of the seronegative individuals later received a nucleic acid amplification test (NAAT) — sometimes referred to as a PCR test — for SARS-CoV-2. The research team looked at what fraction of individuals in each group subsequently had a positive NAAT result, which may indicate a new infection. The study team reviewed NAAT results at several intervals: 0-30 days, 31-60 days, 61-90 days, and >90 days because some people who have recovered from a SARS-CoV-2 infection can still shed viral material (RNA) for up to three months (although they likely do not remain infectious during that entire period). The team found that, during each interval, between 3% and 4% of the seronegative individuals had a positive NAAT test. But among those who had originally been seropositive, the NAAT test positivity rate declined over time. When the researchers looked at test results 90 or more days after the initial antibody test (when any coronavirus detected by NAAT is likely to reflect a new infection rather than continued virus shedding from the original infection), only about 0.3% of those who had been seropositive had a positive NAAT result — about one-tenth the rate in those who had been seronegative. Although these results support the idea that having antibodies against SARS-CoV-2 is associated with protection from future infection, the authors note important limitations to this study. In particular, the findings come from a scientific interpretation of real-world data, which are subject to biases that may be better controlled for in a clinical trial. For example, it is not known why people who had tested antibody positive went on to have a PCR test. In addition, the duration of protection is unknown; studies with longer follow-up time are needed to determine if protection wanes over time. To continue to comprehensively address this important research question, NCI is supporting clinical studies that monitor infection rates in large populations of people whose antibody status is known. These are known as "seroprotection" studies. NCI is also sponsoring ongoing studies using real-world data to assess the longer-term effect of antibody positivity on subsequent infection rates. This research is part of a $306 million effort that NCI has taken on at the request of Congress to develop, validate, improve, and implement serological testing and associated technologies applicable to COVID-19. Through this appropriation, NCI is working with the Department of Health and Human Services; the National Institute of Allergy and Infectious Diseases, another part of NIH; and other government agencies to apply its expertise and advanced research capabilities to respond to this pandemic, including efforts to rigorously characterize the performance of serology assays. About the National Cancer Institute (NCI): NCI leads the National Cancer Program and NIH's efforts to dramatically reduce the prevalence of cancer and improve the lives of cancer patients and their families, through research into prevention and cancer biology, the development of new interventions, and the training and mentoring of new researchers. For more information about cancer, please visit the NCI website at cancer.gov or call NCI's contact center, the Cancer Information Service, at 1-800-4-CANCER (1-800-422-6237). About the National Institutes of Health (NIH): NIH, the nation's medical research agency, includes 27 Institutes and Centers and is a component of the U.S. Department of Health and Human Services. NIH is the primary federal agency conducting and supporting basic, clinical, and translational medical research, and is investigating the causes, treatments, and cures for both common and rare diseases. For more information about NIH and its programs, visit www.nih.gov. NIH…Turning Discovery Into Health® |
| You are subscribed to email updates from "infection inc" - Google News. To stop receiving these emails, you may unsubscribe now. | Email delivery powered by Google |
| Google, 1600 Amphitheatre Parkway, Mountain View, CA 94043, United States | |



Comments
Post a Comment